A Hidden Markov Model (HMM) is a stochastic model that captures the
statistical properties of observed real world data. A good HMM accurately
models the real world source of the observed data and has the ability to
simulate the source. Machine Learning techniques based on HMMs have been
successfully applied to problems including speech recognition, optical
character recognition, and as we will examine, problems in computational
biology. The main computational biology problems with HMM-based solutions
are protein family profiling, protein binding site
recognition and the problem that is the topic of this paper, gene finding
in DNA.
Genefinding or gene prediction in DNA has become one of the foremost
computational biology problems for two reasons. First, because completely
sequenced genomes have become readily available. And most importantly,
because of the need to extract actual biological knowledge from
this data to explain the molecular interactions that occur in cells and to
define important cellular
pathways. Discovering the location of genes on the genome is the
first step towards building a such a body of knowledge. This paper will
introduce several different statistical and
algorithmic methods for gene finding, with a focus on the statistical
model-based approach using HMMs.
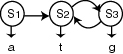
A basic Markov model of a process is a model where each state corresponds
to an observable event and the state transition probabilities depend only
on the current and predecessor state.
This model is extended to a Hidden Markov model for application to more
complex processes, including speech recognition and computational
genefinding. A generalized Hidden Markov Model (HMM) consists of a finite
set of states, an alphabet of output symbols, a set of state transition
probabilities and a set of emission probabilities. The emission
probabilities specify the distribution of output symbols that may be
emitted from each state.
Therefore in a hidden model, there are two stochastic processes; the
process of moving between states and the process of emitting an output
sequence. The sequence of state transitions is a hidden process and is
observed through the sequence of emitted symbols.
Let us formalize the definition of an HMM in the following way, taken from an HMM tutorial by Lawrence Rabiner [14]. An HMM is defined by the following elements:
aij = P(qt+1 = Sj | qt = Si), 1 È i,j È N
bj(k) = P(vk | qt = Sj), 1 È j È N, 1 È k È M.
pi = P(q1 = Si), 1 È i È N.
Once the architecture of an HMM has been decided, an HMM must be trained
to closely fit the process it models. Training
involves adjusting the transition and output probabilities until the model
sufficiently fits the process. These adjustments are performed using
standard machine learning techniques to optimize P(O | l), the
probability
of observed sequence O = O1 O2 ... OT, given model l over a
set of training sequences.
The most common and straightforward algorithm for HMM
training is expectation maximization (EM)1 which adapts the transition and
output parameters by continually reestimating these parameters until P(O | l) has been locally maximized.
HMM decoding involves the prediction of hidden states given an observed
sequence. The problem is to discover the best sequence of states Q = q1q2 ... qT visited that accounts for an emitted sequence O = O1 O2 ... OT
and a model l. There may be several different ways to define
a best sequence of states. A common decoding algorithm is the Viterbi
algorithm2. The Viterbi algorithm uses a
dynamic programming approach to find the most likely sequence of states
Q given an observed sequence O and model l.
The field of computational biology involves the application of computer
science theories and approaches to biological and medical problems.
Computational biology is motivated by newly available and abundant raw
molecular datasets gathered from a variety of organisms. Though
the availability of this data marks a new era in biological research, it
alone does not provide any biologically significant knowledge. The goal of
computational biology is then to elucidate additional information regarding
protein coding, protein function and many other cellular mechanisms from
the raw datasets. This new information
is required for drug design, medical diagnosis, medical treatment and
countless fields of research.
The majority of raw molecular data used in computational biology
corresponds to sequences of nucleotides corresponding to the primary structure
of DNA and RNA or sequences of amino acids corresponding to the primary
structure of proteins. Therefore the problem of inferring knowledge from
this data belongs to the broader class of sequence analysis problems.
Two of the most studied sequence analysis problems are speech recognition
and language processing. Biological sequences have the same left-to-right
linear aspect as sequences of sounds corresponding to speech and sequences
of words representing
language. Consequently, the major computational biology sequence analysis problems
can be mapped to linguistic problems. A common linguistic metaphor
in computational biology
is that of protein family classification as speech recognition. The
metaphor suggests interpreting
different proteins belonging to the same family as different vocaIizations
of
the same word. Another metaphor is gene finding in DNA sequences as the
parsing of language into words and semantically meaningful sentences. It
follows that biological sequences can be
treated linguistically with the same techniques used for speech
recognition
and language processing.
Since the theory of HMMs was formalized in the late 1960s, several
scientists have applied the theory to speech recognition and language
processing.
Just as HMMs were first introduced as mathematical models of
language, HMMs can be used as mathematical models of molecular processes
and biological sequences. In addition, HMMs have been applied to
linguistics because
they are suited for problems where the exact theory may be unknown but where there exists large amounts of data and
knowledge derived from observation. As this is also the situation in
biology,
HMM-based approaches have been successfully applied to problems in
computational biology. The main benefit is that an HMM provides a
good method for learning the theory from the data and can provide a
structured model of sequence data and molecular processes.
It is clear that an HMM-based approach is a logical idea for tackling
problems in computational biology. Much work has been performed and
applications have been built using such an approach. Baldi and Brunak [1]
define three main
groups of problems in computational biology for which HMMs have been
proven
especially useful.
First, HMMs can be used for multiple alignments of DNA sequences, which is a difficult task to perform using a naive dynamic programming approach. Second, the structure of trained HMMs can uncover patterns in biological data. Such patterns have been used to discover periodicities within specific regions of the data and to help predict regions in sequences prone to forming specific structures. Third is the large set of classification problems. HMM based approaches have been applied to structure prediction - the problem of classifying each nucleotide according to which structure it belongs. HMMs have also been used in protein profiling to discriminate between different protein families and predict a new protein's family or subfamily. HMM-based approaches have also been successful when applied to the problem of gene finding in DNA. This is the problem of classifying DNA bases according to which type of job they perform during transcription. The remainder of this paper is concerned with this last problem of computational gene finding.
The problem of finding genes in DNA has been studied for many years. It was one of the first problems tackled once sufficient genomic data became available. The problem is given a sequence of DNA, determine the locations of genes, which are the regions containing information that code for proteins. At a very general level, nucleotides can be classified as belonging to coding regions in a gene, non-coding regions in a gene or intergenic regions. The problem of gene finding can then be stated as follows:
Input: A sequence of DNA X = (x1,...xn) ö S*, where S = A,C,G,T.
Output: Correct labeling of each element in X as a belonding to a coding region, non-coding region or intergenic region.
Gene finding becomes complicated when the problem
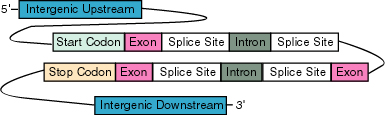
is approached in more biological detail. A eukaryotic gene contains coding regions called exons which may
be interupted by non-coding regions called introns. The exons and introns are
separated by splice sites. Regions outside genes are called intergenic. The
goal of gene finding is then to annotate the sets of genomic data with the location of genes
and within these genes, specific areas such as promoter regions, introns
and exons.
Many effective tools based on HMMs have been created for the purpose of gene finding. Among the most successful tools are Genie, GeneID and HMMGene [8]. Though each tool has a slightly different model, they each use the technique of combining several specialized submodels into a larger framework. The submodels correspond directly to different regions of DNA defined according to their function in the process of gene transcription. Most of the gene finding tools are hybrid models that include neural network components. In these tools, instead of an HMM, a neural network models certain regions, such as splice sites [12]. The overall framework of an HMM-based gene finder combines the submodels into a larger model corresponding to the organization of a gene in DNA and its functional roles. The following figure shows the different regions of DNA separated according to their function in transcription and translation.
The HMM-based approach to gene finding has been well studied and well implemented. There are many tools available online, comprehensive analyses available and a comparison of the tools on a benchmark dataset by Burset and Guigo [2]. An extensive list of HMM-based gene finding resources is contained in [8]. Currently, gene finding tools are very good at classifying regions as coding or non-coding, but are not yet able to accurately predict the exact boundaries of exons. There are also complicated situations which cause poor gene finding performance such as multiple exon genes, nested genes and unusual genes. New work in gene finding applications involves the utilization of information contained within the data sets of known genes. With a large number of known genes in many organisms, there is a high chance that a newly discovered gene will be related to an already known gene. Therefore, gene finding can be attempted by matching new sequences of DNA against sets of known genes.
My goal in this project is to learn about HMMs and their applications to problems in computational biology. In order to gain a strong understanding of the algorithms involved, I had plans to implement an HMM to perform gene finding. As my research progressed3 I decided to learn about HMM algorithms by studying a simplified subproblem of gene finding. I chose to build and compare two HMMs for identify coding and non-coding regions in genes.
The following section briefly describes the HMM algorithms I used in this experiment. For a complete explanation of these algorithms
and their correctness, see [14]. The algorithms and
notations used below follow directly from [6].
The basic task of building an HMM for a specific problem is to infer a set of parameters q for a model l from a set of data D. For simplicity of notation, assume that there is one sequence in the training set and D is a sequence of values d1, ..., dL. In this experiment, the problem is a classification problem and therefore each data element di can be represented as a value xi and its corresponding class label yi. D is then (X,Y) where X = x1, ..., xL be the sequence of values and Y = y1, ..., yL be the corresponding sequence of class labels. With respect to the problem of distinguishing coding and non-coding regions in DNA, X is a sequence of DNA and Y is a sequence identifying each DNA base as belonging to a coding region or non-coding region.
I implement two different strategies for performing the inference of HMM parameters for this problem. The first is the Maximum Likelihood (ML) approach, which infers the parameters by maximizing P(X, Y | l), the probability of the data set over all parameter sets for model l.
The ML approach to modeling a classification problem involves combining
several separately trained submodels into a larger model framework. Each
submodel corresponds to a distinct class and is trained with data belonging to
its respective class. It follows that all the elements of a training
sequence for a specific
submodel share the same class label.
The second is the Conditional Maximum Likelihood (CML) approach, which infers the parameters by maximizing P(Y | X, l), the probability of the labeling over all parameter sets for model l.
The CML approach does not train submodels separately. Instead, it trains the entire model with a single class-labeled dataset.
Expectation maximization (EM) is the standard algorithm for maximizing
the HMM parameters when the paths through the model for each training
sequence are unknown. The standard EM algorithm is the Baum-Welch
algorithm. This iterative algorithm has two steps, the expectation step
and the maximization step. First, it calculates the expected number of times each
transition and emission is used for the training set. Then, the transition
and emission parameters are updated using reestimation formulas.
Recall the definition of HMM elements from the beginning of the paper. For ML and CML, the reestimation formulas are:
[`(p)]i = expected number of times in state Si at time t = 1.
[`a]ij = expected number of transitions from state Si to state Sj / expected number of transitions leaving state Si
[`b]j(k) = expected number of times in state Sj and observing symbol vk / expected number of times in state Sj
The expectation values are calculated using quantities determined in the
forward and backward algorithms. The forward algorithm determines
fk(i) = P(x1 ... xi, qi = Sk | l), the probability of the observed
sequence up to and including the ith term and being at state Sk at
time i. The backward algorithm is very similar. It determines bk(i) = P(xi+1 ... xL | qi = Sk, l), the probability of observing
sequence from i+1 to the end given that the state at time i is Sk.
For the complete forward and backward recusions and algorithms see
[14].
ML and CML differ in the way that the expected values are tabulated. Though they both use the forward and backward tems, only correctly labeled paths are considered in CML. So, the forward and backward terms for states l that emit a different label than the observed label yi are fl(i) = 0 and bl(i) = 0.
I constructed a very simple model structure of 8 states.
The model consists of two submodels corresponding to
coding and non-coding regions respectively. This model is oversimplified
and to be scientifically useful, as it should include states corresponding to
promoter regions and splice sites.
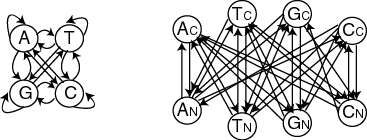
Using a so labeled architecture means that the emission probabilities did not have to be learned. This is often the approach taken when learning simple probabilistic properties of DNA sequences, but would not be effective in larger models, such as those used for gene finding.
I downloaded HMM algorithms from [10] and rewrote them to deal with training sets with multiple sequences. I also wrote code to implement the CML approach described above by ensuring that only the desired class paths are considered when determining the reestimation values.
The datasets used were created from publicly available coding and non-coding vertebrate sequence data [2]. I created 5 sets of coding data, with each set containing 50 vertebrate coding sequences of length 300. I also created 5 sets of non-coding data, with each set containing 50 vertebrate non-coding sequences of length 350. The test data is a random combination of coding and non-coding sequences not seen during model training. The measure of goodness of each model is determined by the percentage of test set bases the model correctly labels.
The main result of my experimenting is that I learned the Baum-Welch and
CML algorithms very well. I also clearly understand the computational
difficulties faced by many bioinformaticians when dealing with large data
sets like genomic sequence sets. Unfortunately, it has become clear that this experiment
is simplified to the point of any measured results have little meaning.
It is very difficult for me to distinguish between random and good
behaviour of the HMMs, as each labeling had a 0.5 chance of being
correct. I also found that though literature suggests the labeled architecture described in the
previous section for such problems [6], it became
very difficult to discern if the parameters were in fact being adjusted to
maximize the probability of the training set given the model.
My incomplete results are quite random numbers, and I did not succeed in fully
training a model using the CML approach. Perhaps the fact that the
models did not convincingly represent the data is because of too little
training data and an oversimplified model. I am convinced that the
expectation and maximization algorithms functioned correctly, as I was
successful in separately training the coding and non-coding submodels. However, I found it
difficult to determine appropriate probabilities to govern the transitions between the
two submodels.
From my newly acquired experience with HMM algorithms and training, I do believe that the CML approach
is well suited to these types of classification problems and
would be effective in a larger framework, as Krogh showed [11].
In conclusion, I believe that though I had difficulty training simple models, HMMs have been, and continue to be, an invaluable tool for infering biologically meaningful information from otherwise nearly incomprehensible DNA sequences.
1 Please see references [1] and [14] for a complete specification of these and other HMM algorithms.
2 As in footnote 1.
3 As I read the material and investigated the available datasets, I realized that gene finding had been done many, many times over and in much greater detail than I would be able to manage before the due date of this project. Also, even the smallest datasets for gene finding are huge! My poor connection at home couldn't cope.