PHYLO_WIN
Phylogenetic Analysiis
PHYLO_WIN reads a set of sequences that have been aligned using programs
such as CLUSTALW or PIMA, and constructs phylogenetic trees using a wide
variety of algorithms. In the example, a group of PR5 (thaumatin/osmotin)
DNA sequneces have been aligned using Pearson's MRTRANS program in GDE.
The aligned sequences were then selected, and Phylogeny -> phylo_win was
selected.
To construct a tree, the Neighbor-Joining method was chosen, and
100 bootstrap replicates were done. The output appears below:
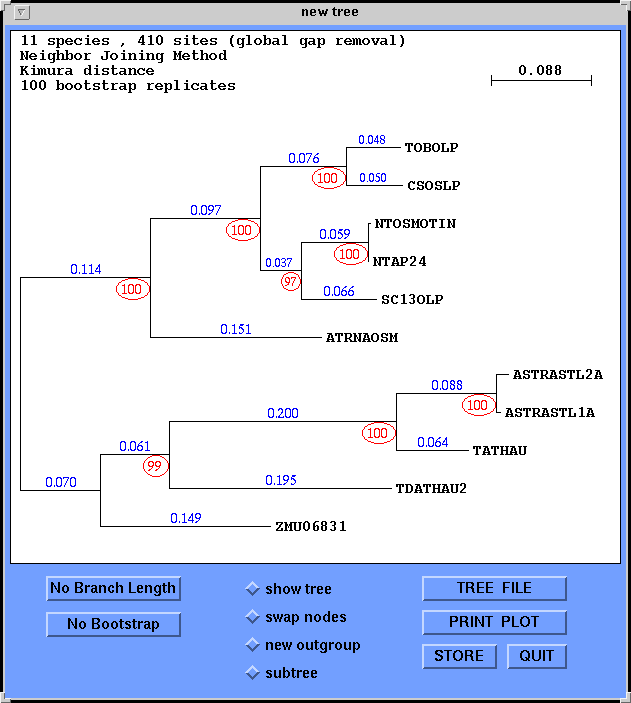
Branch lengths are indicated in blue. Red circled numbers indicate
the number of times a branch appeared out of 100 bootstrap replicates.
Trees can be saved to a file by choosing TREE FILE. A PostScript file of
the tree can be generated using PRINT PLOT.
A full online-help is available.
PHYLO_WIN is most convenient to run from GDE. Select three or more
aligned protein or DNA sequences and choose Phylogeny -> phylo_win. PHYLO_WIN
can also be run from the command line by typing 'phylo_win'. PHYLO_WIN
can read aligned sequences in CLUSTAL, FASTA, MASE, NEXUS-PAUP, and PHYLIP
formats.
The phylo_win website is at http://pbil.univ-lyon1.fr/software/phylowin.html